events
enjoying respectful, inclusive events
Thank you for participating in an Allen Institute event.
Events are central to our mission of scientific discovery and our commitment to big, team, and open science. We are dedicated to creating environments—both in person and virtual—where all participants feel safe, respected, and welcomed.
All attendees, speakers, organizers, and staff are expected to uphold this Code of Conduct and contribute to a positive, inclusive experience for everyone.
Events are central to our mission of scientific discovery and our commitment to big, team, and open science. We are dedicated to creating environments—both in person and virtual—where all participants feel safe, respected, and welcomed.
All attendees, speakers, organizers, and staff are expected to uphold this Code of Conduct and contribute to a positive, inclusive experience for everyone.

Summer Workshop on the Dynamic Brain 2026
The 2026 Summer Workshop on the Dynamic Brain is an intensive, project-based residential course with a focus on the neurobiology of sensory processing, coding, and neural population dynamics.

explore all events
Thank you! Your submission has been received!
Oops! Something went wrong while submitting the form.

Coding with Allen Institute Data: A Python Workshop for Undergraduate Educators 2026
This workshop will introduce undergraduate neuroscience educators to working with Allen Institute open datasets using Python.
Brain Science
Neural Dynamics
Educators
Exploring Cell Types with the Allen Institute - Buenos Aires 2026
This free, two-day workshop in Buenos Aires focused on open access single-cell neuronal and non-neuronal transcriptomic data and tools from the Allen Institute.
Brain Science
Postdocs
Scientists
Early Career Scientists
Exploring Cell Types with the Allen Institute - Boston 2026
This free, full-day workshop in Boston focuses on how brain cell types are defined, cataloged, and explored—and how to use the Allen Institute’s open data and tools in your own research.
Brain Science
Postdocs
Scientists
Early Career Scientists
Summer Workshop on the Dynamic Brain 2026
The 2026 Summer Workshop on the Dynamic Brain is an intensive, project-based residential course with a focus on the neurobiology of sensory processing, coding, and neural population dynamics.
Brain Science
Graduate
Scientists

Cascadia Mucosal Biology Symposium
The inaugural Cascadia Mucosal Biology Symposium hosted by the University of Washington and the Allen Institute in Seattle, Washington, September 10 - 11, 2026.
Immunology
Early Career Scientists
Postdocs
Scientists
Students

Stem Cell & Developmental Biology Early Career Symposium
For the first time, the International Society for Stem Cell Research (ISSCR), the Society for Developmental Biology (SDB), and the Allen Institute for Cell Science (AICS) are collaborating to present a three-day scientific symposium led by early-career scientists.
Cell Science
Scientists

2026 Lake Conference – Comparative and Evolutionary Neurobiology
Join the Allen Institute and Circuit Neuroscience Basel for the Lake Conference in Seattle, Washington this October.
Brain Science
Neural Dynamics
Postdocs
Scientists
Graduate
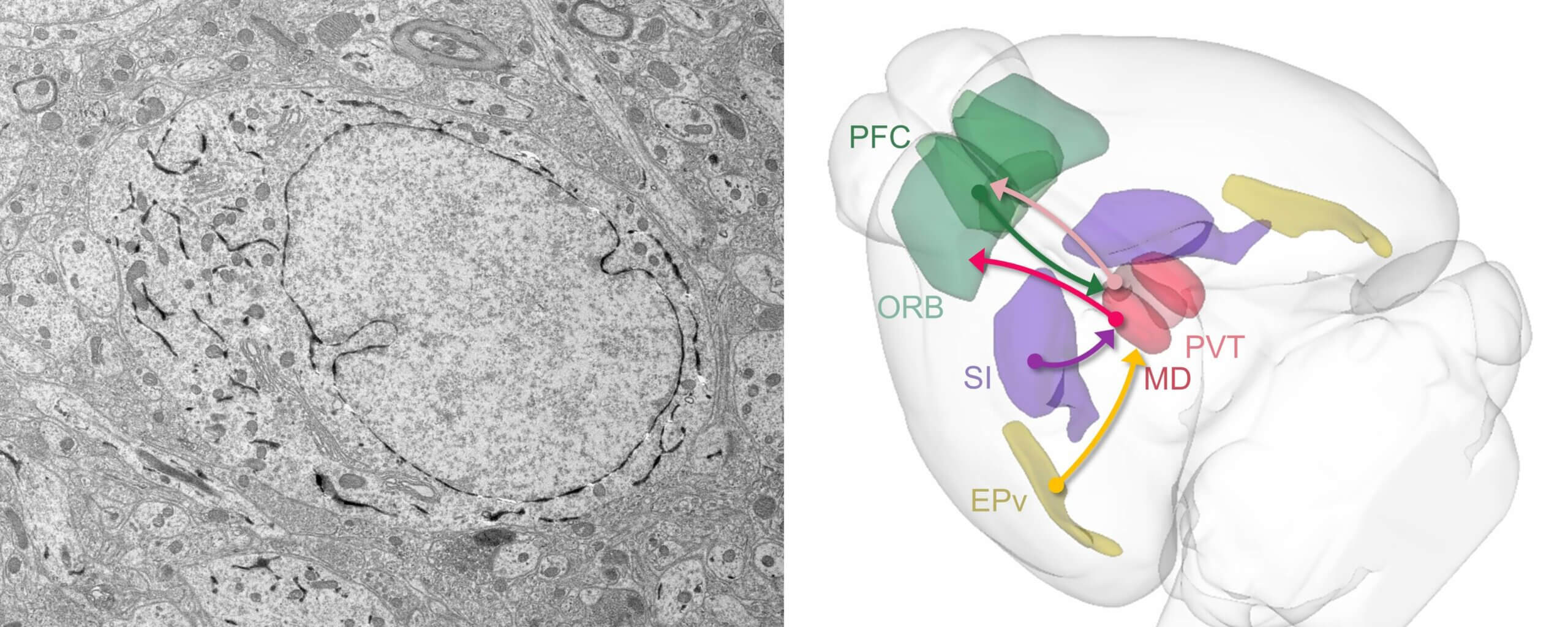
Emerging connectomics of thalamus and striatum workshop
Join us for a hands-on opportunity to learn how to use volumetric electron microscopy (EM) data and address biological questions central to the thalamus and striatum at the Emerging Connectomics of the Thalamus and Striatum Workshop, hosted at by the Allen Institute.
Neural Dynamics
Graduate
Postdocs
Scientists